Zdravotnícke pomôcky triedy I - informácie pre výrobcov so sídlom v SR
Obsah
ZDRAVOTNÍCKE POMÔCKY TRIEDY I - INFORMÁCIE PRE VÝROBCOV SO SÍDLOM V SR
Návod pre výrobcov zdravotníckych pomôcok triedy I so sídlom v SR
Výrobcovia zdravotníckych pomôcok (ZP) triedy I, ktorí majú v úmysle uviesť na trh tieto ZP, by mali chronologicky postupovať nasledovne:
1. Potvrdiť, že výrobok je ZP, t.j. vyhovuje definícii ZP uvedenej v článku 2 nariadenia európskeho parlamentu a rady o zdravotníckych pomôckach č. 2017/745 (ďalej len „nariadenie č. 2017/745“) , v prípade hraničného výrobku požiadať o konzultáciu ŠÚKL.
2. Potvrdiť, že ide o ZP triedy I v súlade s pravidlami triedenia uvedenými v prílohe VIII nariadenia č. 2017/745, pričom, ak sa dajú použiť viaceré pravidlá, musí sa použiť najprísnejšie z nich. Pri klasifikácii je dôležité zohľadniť tieto aspekty: účel určenia, dĺžku použitia, miesto použitia na ľudskom tele, či je ZP aktívna alebo nie, či je ZP invazívna alebo neinvazívna a pod.
3. ZP musia spĺňať základné požiadavky uvedené v prílohe I nariadenia 2017/745, ktoré sa na ne vzťahujú, s prihliadnutím na účel určenia. ZP musia byť navrhnuté a vyrobené takým spôsobom, aby pri použití za normálnych podmienok a na účely určené výrobcom, neohrozili klinický stav a bezpečnosť pacienta, ani bezpečnosť a zdravie používateľov alebo iných osôb za predpokladu, že všetky riziká, ktoré môžu byť spojené s ich použitím sú prijateľnými rizikami v porovnaní s prínosom pre pacienta a zodpovedajú vysokej úrovni ochrany zdravia a bezpečnosti. Zariadenie musí dosiahnuť výkon, aký bol určený výrobcom.
4. Výrobca musí pripraviť technickú dokumentáciu. Výrobca alebo splnomocnený zástupca v EÚ musí mať technickú dokumentáciu, ktorá preukazuje zhodu jeho výrobkov s požiadavkami nariadenia č. 2017/745. Táto technická dokumentácia musí byť pripravená pred vypracovaním ES vyhlásenia o zhode, aby bola k dispozícii na posúdenie príslušnému orgánu v SR - ŠÚKL. Výrobcovia by mali preveriť jazykové požiadavky u príslušného orgánu štátu, kam zo SR exportujú ZP. Technická dokumentácia by mala byť pripravená z prehľadu základných požiadaviek a ďalších príslušných požiadaviek smernice a musí zahŕňať aspekty podľa prílohy II nariadenia č. 2017/745:
5. Výrobca požiada notifikovanú osobu o účasť pri posudzovaní zhody, ktorá je v prípade ZP triedy I obmedzená iba na:
aspekty výroby, ktorými sa dosiahnu sterilné podmienky v prípade ZP triedy I sterilných (postup opísaný v prílohe IX alebo XI nariadenia č. 2017/745),
aspekty výroby spojené so zhodou výrobkov s metrologickými požiadavkami v prípade ZP triedy I s meracou funkciou (postup opísaný v prílohe IX alebo XI nariadenia č. 2017/745),
opakovane použiteľnými chirurgickými pomôckami triedy I (postup opísaný v prílohe IX alebo XI nariadenia č. 2017/745).
V ostatných prípadoch sa účasť notifikovanej osoby nevyžaduje, je mimo regulačného rámca nariadenia č. 2017/745.
6. Výrobca pripraví návod na použitie a samotné označenie ZP – štítok. Ku každej ZP sa musia priložiť informácie potrebné na jej bezpečné použitie a na identifikáciu výrobcu alebo splnomocneného zástupcu v EÚ s prihliadnutím na vzdelanie a vedomosti potenciálnych používateľov. Táto informácia sa skladá z označenia (štítku) a údajov v návode na použitie. Návod na použitie nie je nutný pre ZP triedy I, ak môžu byť bezpečne používané aj bez takéhoto návodu, napr. obväzy, okuliare, vychádzkové palice a iné.
Národné jazykové požiadavky musia byť brané do úvahy vo vzťahu k označeniu ZP a návodu na použitie. Používané jazykové verzie majú byť zahrnuté do technickej dokumentácie.
Vzhľadom na technický pokrok v informačných technológiách a zdravotníckych pomôckach by malo byť umožnené poskytnutie takýchto informácií (návodu na použitie) výrobcom aj iným spôsobom, napr. elektronicky.
Návod na použitie v slovenskom jazyku musí obsahovať podľa potreby údaje v súlade s prílohou I kapitoly III ods. 23.4 nariadenia č. 2017/745. Označenie zdravotníckej pomôcky musí obsahovať podľa potreby údaje v slovenskom jazyku v súlade s prílohou I kapitoly III ods. 23.2. nariadenia č. 2017/745.
7. Výrobca vypracuje ES vyhlásenie o zhode. ES vyhlásenie o zhode je postup, ktorým výrobca alebo splnomocnený zástupca v EÚ zabezpečuje a vyhlasuje, že príslušné výrobky spĺňajú ustanovenia nariadenia č. 2017/745, ktoré sa ich týkajú. ES vyhlásenie o zhode by malo obsahovať všetky informácie potrebné na určenie smerníc, podľa ktorých bolo vypracované, rovnako ako informácie o výrobcovi, splnomocnenom zástupcovi v EÚ, notifikovanej osobe a ZP, prípadne odkaz na harmonizované normy alebo iné relevantné dokumenty. ES vyhlásenie o zhode obsahuje minimálne informácie uvedené v prílohe IV nariadenia č. 2017/74
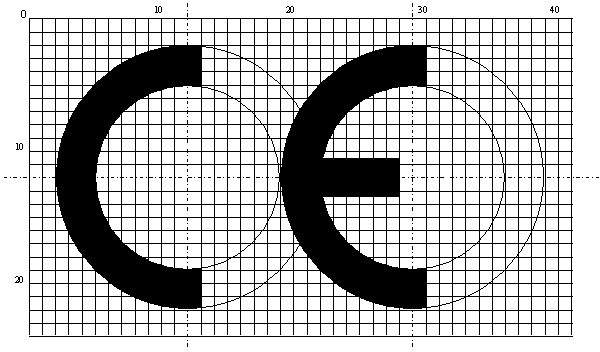
8. Výrobca označí ZP značkou zhody CE a dodrží jej proporcie podľa rastrovanej kresby, pričom jej vertikálny rozmer nesmie byť menší ako 5 mm. Všetky ZP triedy I uvedené na trh musia byť označené značkou zhody CE, ktorá musí byť pripojená v dobre viditeľnej, čitateľnej a nezmazateľnej forme na ZP, jej sterilnom obale - pokiaľ je to možné a vhodné, ďalej v návode na použitie, rovnako ako na každom predajnom balení. V prípade ZP triedy I uvedených na trh v sterilnom stave a/alebo ZP triedy I s meracou funkciu, opakovane použiteľnými chirurgickými pomôckami triedy I musí byť za alebo pod označením CE uvedené identifikačné číslo notifikovanej osoby. Je zakázané pripájať na ZP značky alebo symboly, ktoré by mohli uviesť do omylu tretie strany, pokiaľ ide o význam značky zhody CE. Ďalšie doplnkové značky môžu byť pripojené na ZP, jej obal, alebo do návodu na použitie, ak viditeľnosť alebo čitateľnosť značky zhody CE nie je zhoršená. V prípade, ak je ZP veľmi malá, môže byť od požiadavky na minimálny vertikálny rozmer značky zhody CE (5 mm) upustené a teda môže byť (po dohode so ŠÚKL) menších rozmerov.
{kind=link}
{kind=link}
9. Oznámenie výrobcu a ZP v ŠÚKL. Výrobca alebo splnomocnený zástupca v EÚ so sídlom v SR oznámi v ŠÚKL seba a ZP triedy I. Viac informácií nájdete tu.
10. Výrobca alebo splnomocnený zástupca v EÚ aktivuje systém vigilancie ZP (medical device vigilance system). Výrobca alebo splnomocnený zástupca v EÚ je zodpovedný za aktiváciu systému vigilancie a musí informovať ŠÚKL o nehodách ZP, info nájdete tu. Po oznámení nehody ZP je výrobca povinný vykonať vyšetrovanie, vypracovať a odoslať ŠÚKL správu z vyšetrovania a v spolupráci so ŠÚKL zvážiť, aké by mali byť prijaté opatrenia. Členský štát, v ktorom prebieha klinické skúšanie ZP musí zaznamenať a oznámiť všetkým príslušným orgánom pre ZP, v ktorých tiež prebieha toto klinické skúšanie ZP, všetky závažné nežiaduce príhody vzniknuté v rámci klinického skúšania.
11. Výrobca preskúma skúsenosti so ZP získané z trhového dohľadu. Výrobca zavedie a bude aktualizovať postup hodnotenia získaných skúseností so ZP uvedenými na trh a bude vykonávať potrebné nápravné opatrenia so zreteľom na charakter a riziko vo vzťahu ku ZP. Každé klinické hodnotenie ZP a technická dokumentácia sa musia aktívne aktualizovať na základe údajov z trhového dohľadu vykonávaného výrobcom po uvedení ZP na trh (post market surveillance).